

原标题:18号染色体及相关疾病

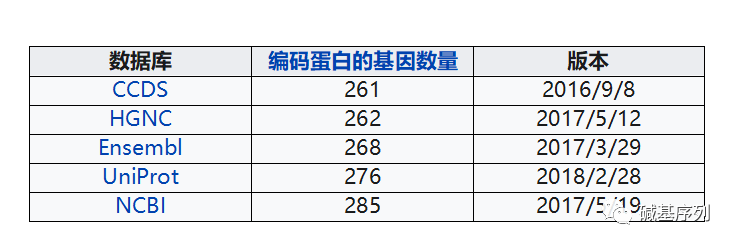
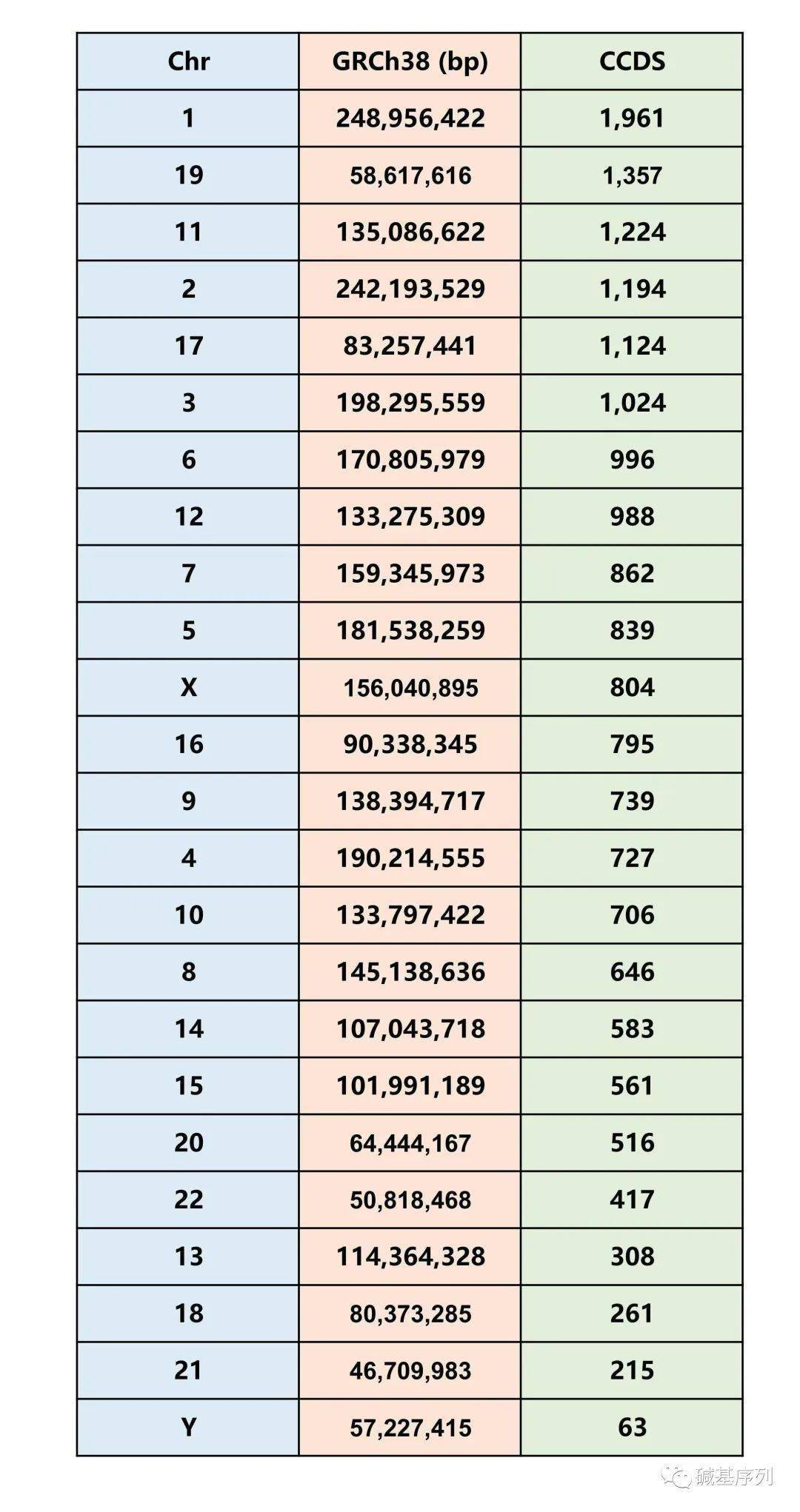
18号染色体是人类的23对染色体之一。在正常的人类体细胞中,会有1对18号染色体,一条来自父亲,一条来自母亲。一条18号染色体有80,373,285 bp (GRCh38),约占人类细胞DNA总长度的2.5%。基因数量261个(CCDS), 倒数第三。
DNA长度
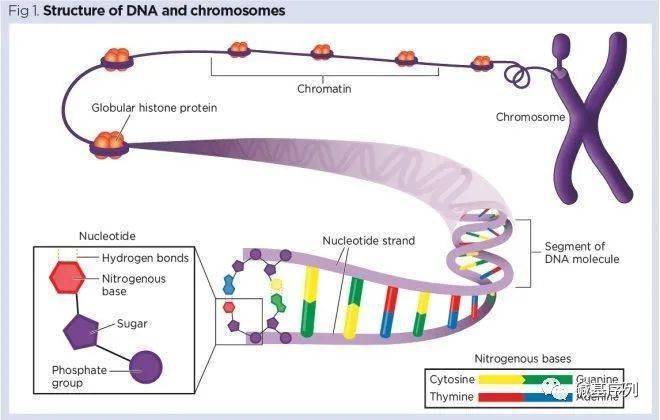
人类的一个细胞中的DNA分子在细胞核中被打包成46条染色体。DNA是一种天然的螺旋分子,进行超卷曲,从而占用更少的空间。利用超螺旋,这样一来,每个细胞中的30亿个碱基对就可以装进只有6微米宽的空间里。如果你把一个细胞里的DNA拼接拉伸到最大,大概有2米长,把人身上所有细胞里的DNA加在一起,长度大约是太阳系直径的两倍。
染色体
染色体(chromosome)来自希腊语 χρῶμα(色度,“颜色”)和 σῶμα(体细胞,“体”),描述了它们对特定染料的强染色。染色体是遗传物质,是基因的载体,人类的常染色体是成对存在的。人体的体细胞染色体数目为23对,其中22对为男女所共有,称为常染色体(autosome);另外一对为决定性别的染色体,男女不同,称为性染色体(sex chromosome),男性为XY,女性为XX。
当细胞不分裂时,染色体在细胞核中是不可见的——在显微镜下也是如此。然而,构成染色体的 DNA 在细胞分裂过程中变得更紧密,染色体在显微镜下可见, 此时我们可以通过显微镜观察染色体的数量和结构,来判断染色体是否正常。
染色体(chromosome)是细胞分裂时DNA 存在的特定形式,DNA 被一种称为 组蛋白的蛋白质紧密卷绕并被包装成一个线状结构。
上图为显微镜下观察到的染色体
上图为染色体模式图
染色体整体的不同部位对颜料的着色能力不同,表现出颜色深浅不一,所以通过显微镜可以观察到每条染色体不同区域的深浅条带,这是区分23条不同染色体的基础。
18号染色体
相关疾病
18号染色体三体
trisomy 18 (T18)
18-三体综合征也称 爱德华兹综合征(Edwards syndrome),是由于18号染色体多一条导致,由爱德华兹于1960年首次报道。T18活产患病率为1/6000~1/8000,产前诊断后胎儿丢失和终止妊娠的频率较高,总患病率较高(1/2500~1/2600)。活产女性患病率高于男性(女性患病人数÷男性患病人数×100%=60.4%),但活产女性的存活率更高。 在绒毛染色体异常导致自然流产中的比例约为3.00%~6.00%。
T18通常是由亲本配子减数分裂或细胞有丝分裂过程中染色体不分裂导致。18号染色体不分离大多发生在 卵子生成减数分裂Ⅱ期(约58.7%)。 完全型T18(约94%)指所有细胞都含有3条完整的18号染色体,额外多出的1条染色体多为母源。
完全型T18通常过期分娩、胎动少、羊水过多、胎盘小及单一脐动脉、出生体重低、肌张力增高、发育迟缓、严重智力障碍。产前超声可发现T18胎儿的特征性临床表现为 “草莓头”,特殊握拳姿势(第3、4指紧贴手掌,第2、5指压在其上),摇椅状足底、心脏畸形、脉络丛囊肿(约1/3)。T18活产儿主要特征性表现包括∶胸骨短、钳 状手和手指弓形纹过多。此外,18-三体综合征会增加肾母细胞瘤和肝母细胞瘤风险。
嵌合型T18( <5%)指同一个体存在2 种不同的细胞系,一种细胞系18号染色体为2个拷贝,另一种细胞系为3个拷贝。这些细胞系来自同一个受精卵,由于染色体不分离导致不同,表型差异非常大,大多患者存活期相对较长。根据既往文献病例报道提示 外周血白细胞或皮肤成纤维细胞中三体细胞的百分比与个体的表型及严重程度或智力水平无相关性;在任何一个个体中,外周血白细胞中三体细胞的百分比与成纤维细胞之间无相关性;嵌合三体细胞组织分布对表型及严重程度影响很大,但因无法获知大脑、生殖器官及其他重要器官的三体细胞比例,仅凭嵌合三体比例无法预测产前表型严重程度及预后。有些表现为完全型T18表型,甚至早期死亡,而有些表型完全正常且可以生育,智力水平也可从严重智力缺陷到智力正常甚至超过平均水平。
Tucker等(2007)研究已报道的33例嵌合型T18患者临床表型及嵌合比例,其中21例女性、12例男性,患者出生时母亲平均年龄为35岁(19~45岁),父亲平均年龄为38岁(22~51岁),外周血18-三体细胞平均比例为53%(2%~100%),皮肤成纤维细胞18-三体细胞平均比例为29%(0~100%)。
嵌合型T18患者表型总结,Tucker等(2007) DOI 10.1002/ajmg.a.31535
T18的产前诊断
嵌合型T18产前可表现出胎动不良(4/8),患儿临床表型主要为出生时身长/体重/头围≤3个百分位数(比例分别为7/13、10/19、4/7),短指(9/16),高腭穹(10/14),小头畸形(9/15),骨龄延迟(4/7),复发性呼吸道感染(5/10)和中耳炎(5/9),室间隔缺损(6/13),第五指弯曲(5/11),小颌畸形(8/19),肌张力减退(7/16),吸吮不良(4/6),发育迟缓(16/28),语言发育迟缓(7/20),青春期延迟(4/10)和卵巢功能早衰(3/8)。除此之外还可表现出异常面容(如浓眉、宽鼻梁、蒜头鼻/短鼻、薄上唇、低位耳、小嘴、高额头),牙齿发育不良,脊柱侧弯,关节过度伸展,色素沉着,肾脏异常,多毛症等,这些表型频率比较低;患者智力水平差异也非常大,43.3%患者智力正常或超过平均水平(13/30),30%为轻度至中度智力低下(9/30),26.7%为重度智力低下(8/30);患者可有正常生育能力,有12例患者年龄超过20岁,其中7例可生育∶4例患者生育过1个孩子为完全型T18,3 例患者共生育5个孩子。并不是所有嵌合型T18患者一出生就被诊断,有些是反复流产或生育过嵌合型T18的孩子才被诊断,其他有些是因为表现出严重精神发育迟滞、畸形特征或其他医学并发症,提示染色体异常,如心脏缺陷被诊断,这也提示多数嵌合型T18患者可长期存活。
嵌合比例与表型异常严重程度关系:Smith等(1999)总结英国及美国研究数据,提示羊水18-三体细胞大于50%的病例中75%(6/8)表现异常,而羊水18-三体细胞小于50%病例中52%(11/21)表现异常。该研究提示随着羊水三体细胞比例增加,胎儿表现异常的风险会增加(妊娠结果正常的羊水三体细胞小于10%,当三体细胞大于31%则未观察到正常活产儿)。
90%以上T18胎儿至少存在一项超声结构异常。产前诊断后约60%~80%的孕妇选择终止妊娠。T18胎儿早产发生率(35%)及分娩期间死亡率(38.5%)高于一般人群。
T18患儿预后差,出生后大多因中枢性呼吸暂停、心力衰竭很快夭折。约50%的18-三体婴儿生存期大于1周,5%~10%的患儿可活至超过1 岁。由于患儿出生后1年生存率低,且存活儿童存在严重的发育障碍,目前最常见的医疗方法是姑息治疗,拒绝任何特定治疗。当T18患者在纯舒适护理下接受治疗时,呼吸暂停和停止治疗为主要死亡原因,当接受强化治疗时,常见死亡原因会发生改变,生存率会增加,例如在日本5%的T18患儿在心脏手术后能活1年。
18号染色体单亲二体
Uniparental disomy
UPD概念在1980年由Eric Engel首先提出,指父或母一方的染色体片段被另一方的同源部分取代,或某一个体的2条同源染色体都来自同一亲本。
单亲异二体(heterodisomy,hetero-UPD):两条染色体来自同一亲本的两条同源染色体。单亲同二体(isodisomy,iso-UPD):两条染色体来自同一亲本的同一染色体。复合型单亲二体(mix-UPD):部分表现为单亲同二体,部分表现为单亲异二体。片段性单亲二体(seg-UPD):染色体的一部分表现为UPD。
UPD机制:
三体自救(trisomy rescue):减数分裂出现错误的精子(卵子)和卵子(精子)结合形成18号染色体三体受精卵,在胚胎早期有丝分裂过程中失去一条染色体。
单体自救(monosomy rescue):出现错误的精子(卵子)和卵子(精子)结合形成18号染色体单体受精卵,在胚胎早期的有丝分裂过程中,染色体只复制不分离而产生的染色体数目正常但是两条染色体均来源于一个亲本的胚胎。单体自救比三体自救更罕见。
有丝分裂重组:在形成受精卵后,由于早期胚胎发生染色单体之间的有丝分裂重组,导致染色体末端会存在嵌合片段性UPD
Liehr创建的UPD数据库(http://cs-tl.de/DB/CA/UPD/0-Start.html)(最新更新时间2022.8.27),统计了每条染色体发生UPD的病例数
18号染色体包含3个印记基因,其中TCEB3C 为母源性表达,与小肠神经内分泌肿瘤相关;另预测BRUNOL4和FAM59A也可能为印记基因,分别为母源性表达和父源性表达(http∶//www.ge-neimprint.com/site/genes-by-species)。
再发风险评估及咨询: 据评估,在新生儿中,UPD发生频率为1:2,000。最常出现在1号、4号、16号、21号、22号和X染色体;普通人群中母源UPD发生率为父源UPD的3倍。疑似遗传疾病人群中,UPD发生率约为2:1,000 (0.2%)。已报道的UPD病例在 6、7、11、14 和 15 号染色体上最常见。由于UPD来源于父/母一方,若父/母17号染色体存在隐性遗传病致病基因突变,则可能导致相应疾病1的发生 。
环状18号染色体
Ring chromosome 18
46,r(18)
环状18号染色体在所有环状染色体中出现比例低,约7%。
环状染色体(Ring chromosomes,RCs)其本质是形成圆形的DNA分子,罕见出现在真核生物核基因组中(约1/50000),主要出现在原核生物、病毒、真核质体和线粒体中。 由于RCs不稳定性,通常不具有遗传性, 大多数环状染色体以新发变异(de novo)为主, 只有1%的病例是遗传的。目前研究发现环形DNA形成主要有三种方式:①染色体两端断裂,断裂处重接形成环形,两个无着丝粒的末端染色体片段丢失,遗传物质的缺失引起相关疾病表型。这种形成可能与紫外辐射有关;另有研究发现遭受切尔诺贝利核灾难的人群,其环状染色体携带者数量增加。②染色体两端粒直接连结成环,这与年龄增加引起的端粒缩短有关,且可遗传。因不涉及遗传物质的缺失,往往无明显临床表型,但可能会有身材矮小、生育生殖问题。③通过染色体片段插入、缺失、重复、重排等复杂变异形成环状。
1962年,Lindsten 等和 Wang 等首次报道了18 号环状染色体。r(18)为首个被鉴定的人类环状染色体。
Carter等(2014)对30例18号环状染色体患者进行了总结,常见的临床表现为神经肌肉异常(肌力减退,小头畸形,白质异常,癫痫等),内分泌及代谢异常(生长激素分泌不足,甲低,新生儿黄疸等),听力异常(听力损失,慢性中耳炎,听觉闭锁等),骨骼畸形(脊柱侧弯,扁平足等),喂养困难(食物反流,吞咽困难等),视力异常(远视斜视,视神经发育不全,散光,弱视等),心脏畸形(肺动脉狭窄,房间隔缺损等),腭裂,呼吸困难等。同时总结了14例18号环状染色体行为∶无法表达想法和有效沟通,无法安全地执行基本生活任务,容易分心,无法集中,社交能力差,无法与他人合作。
18号环状染色体患者具体的症状取决于18号染色体成环过程中断裂的位置、缺失/重复的大小、涉及的基因以及是否有次级畸变等。
18q21.2的TCF4基因,该基因点突变或缺失可导致 皮特霍普金斯综合征,临床表现为中度至重度智力运动障碍,呼吸急促,抽搐癫痫以及独特的面部特征,包括薄眉毛,凹陷的眼睛,突出的鼻梁,高鼻梁,上唇明显的双曲线(丘比特的弓),宽嘴唇,嘴唇丰满,间距宽牙齿,厚杯状耳朵。 环状染色体缺失不伴有TCF4基因缺失则临床表现为轻度智力障碍。
柴乐等(2022)报道一个患儿,女,11岁7个月,因身材矮小于内分泌科就诊。查体∶身高1.20m,特殊面容∶耳位低,招风耳,牙齿排列不整齐。相关检查心肌酶谱高,肝脏血管瘤,松果体囊肿,并且存在尿失禁的症状,智力低于同龄人。患者核型46,XX,r(18)(p11.3q23)[70]/46,XX,der(18)r(18)(18)(p11.3q2 3;p11.3q23)[14]/45,XX,-18[6]。
张一宁等(2014)报道了1例伴生长激素缺乏的18号环状染色体综合征,患儿身材矮小,有特殊面容,体质量增长缓慢,语言能力较弱,韦氏智力量表测试为轻度智能发育迟缓,行染色体核型分析后,结果为∶46, XX, +r(18),结合临床表现及检查结果,患儿诊断为生长激素缺乏并18号环状染色体综合征。
张秀玲等(2006)报道了1例18号环状染色体的病例,患儿具特殊容貌,身材矮小,无家族遗传病史,脑部发育落后,垂体发育不良,智力低于正常,染色体核型分析结果为∶46, XX, r(18)(p11.2q23)。
18q 缺失综合征
18q部分缺失综合征通常是指18q远端缺失,其发生率高于近端缺失,绝大部分18q部分缺失不具有一致的断裂点,即缺失片段的位置及长度均不固定。在活产儿中发生率约为1/40000。此病患者女性较多,发病率与年龄无关,其中80%病例是新发缺失,10%是由亲代臂间倒位或易位所致。
18q缺失综合征患者的临床表型差异较大,主要临床表现为智力障碍,全身多处发育不全,小头,声音低哑,75%患儿有大而宽翻翘的嘴唇或鲤鱼嘴,50%发生外耳道闭锁、唇裂、腭裂、斜视或眼球震颤,1/3有先天性心脏疾患、马蹄肾,男性有隐睾症,指纹多为涡状纹等。
相当一部分18q部分缺失的病例的断裂点位于18q22.1区域上约1.65Mb的范围内,处于SERPINB8和CDH7基因之间的基因组区域,因此其遗传异质性较高。
一些学者通过比较具有同一表型特征的病例的18q部分缺失的片段大小和断裂点差异,从而定位特定表型的关键区域,如脑白质异常、腭裂、肾脏异常、先天性心脏病、足畸形及IgA缺乏等疾病的关键区域为18q22.3-q23、身材矮小或生长激素缺乏关键区域为18q12.1-q12.3、18q21.1-q21.33及18q22.3-q23、小头畸形关键区域为18q21.33等。
18p 缺失综合征
18p部分缺失综合征是一种由于18 号染色体短臂全部或部分缺失所导致的疾病,呈常染色体显性遗传,在活产婴儿中发病率约为1/50 000。患儿多数为新发生的染色体畸变。在~85%已发表的病例中,18p缺失是新发突变。Hasi-Zagoj等(2015)对 56名新发缺失患者的研究,发现其中25人缺失是父亲的染色体片段。
18p缺失患者常见的表型包括轻到重度智障、语言发育落后、身材矮小、特殊面容、短颈蹼颈,骨骼畸形包括脊柱侧凸/后凸、漏斗胸、髋内翻、髋关节脱臼、足部畸形、 严重畸形有先天性心脏病或脑畸形。少见的表型包括行为异常和自身免疫性疾病。
强敏等(2016)报道了1例18p11.32-p11.21(14.963 Mb)缺失的6月龄女婴,其临床表型主要为肌张力减退、面容异常;18p11.32-p11.21区域的缺失可导致面容异常、行为发育异常等,随着年龄的增长可导致轻-重度智力障碍等,其致病性在不同生长发育阶段可出现不同异常表型。
陈雪君等(2021)报道一例,孕妇26周岁,孕12周,无创产前检测提示18号染色体p11.32-p11.31片段部分(1~6Mb)缺失;孕22周产前超声检查提示∶子宫内单活胎,头位,胎儿发育各测量值正常,羊水适中。胎儿羊水 CMA 检测结果为18号染色体p11.32区域存在6.66Mb片段缺失;孕妇外周血的CMA检测结果与胎儿羊水结果一致,丈夫外周血CMA检测结果正常。孕期胎儿超声各项指标未见异常,胎儿头颅磁共振未检查,电话随访时得知 胎儿孕33周时停止发育,引产后外观表型未见明显异常。 推测引起胚胎停止发育原因可能与胎儿TGIF1基因缺失导致颅脑发育、体内脏器发育畸形相关。
18p11.32-p11.31区域覆盖TGIF1(602630)、LPIN2(605519)、SMCHD1 (614982)3个 OMIM 致病基因, 18p11.31区域也被定义为4型前脑无裂畸形关键区域。TGIFl 对单倍体剂量不足敏感, TGIF基因编码转化生长因子-β诱导因子,属于进化上保守的、非典型的同源结构域蛋白质家族。TGIF水平降低,会增强视黄醇类X受体与类视色素-应答启动子的结合,导致类维生素A调控基因的活动过度和模拟会改变Nodal/TGF-信号传导通路,从而导致前脑无裂畸形,因此该基因变异或缺失可导致前脑无裂畸形4型和垂体发育异常。 在18p 缺失综合征中仅10%的患者表现为前脑无裂畸形。存在TGIFl缺失的18p缺失综合征患者中前脑无裂畸形表型发生率低。 由于 TGIF基因表现为常染色体显性遗传模式而外显率低导致外显不全。
生长激素缺乏在18p缺失的病例中比较常见,可以考虑生长激素的治疗。已有两例18p 缺失的病例接受生长激素治疗有效的报道,18p缺失的病例可以考虑进行生长激素检测,其中生长激素缺乏患者可以给予生长激素治疗。
18q重复
1972年,Cohen等首次报道 18q 部分三体。单纯18q的末端重复并不多见, 可源自新发突变或平衡易位携带者,多数是由不平衡重组导致。 Henson等(2012)分析了已报道的单纯18q11.2-qter重复患者的临床表型,发现部分18q三体通常表现为不同程度的智障、运动发育落后、生长迟滞、癫痫以及特殊面容。 不同重复区域的部分18q三体表型从轻度到重度均有报道,一些病例可以表现为18三体的特征。
18q21.1-qter重复,其临床表型特点为∶女性发病率高于男性,畸形较轻,特殊面容,长头畸形,前额高,发际线高,小下颌,小睑裂,硬腭高拱,鼻短,球状鼻尖,皮肤皱褶增多,低位耳,窄而扁平胸,疝气,掌大而手指短,脚趾畸形,肌张力增高,内脏畸形罕见,生长轻度受限,无新生儿早期死亡,智力存在个体差异,生殖器畸形少见。
在18号染色体长臂上存在3个重要区域∶q11为完全型18-三体综合征的关键性区域;q12.1-q12.3异常只会导致轻微的表型异常;q21.1-qter为18qter综合征的决定性区带。
典型的爱德华综合征的临床表型与18号染色体长臂上的 q12.1→q21.2及 q22.3→qter这两个关键区域三倍体重复的协同作用相关,而重度精神发育迟缓则与q12.1→q21.2的三倍体重复相关。
刘敏等(2018)报道一例胎儿(20周) 染色体核型分析为46,XX,dup(18)(q21-23),其表型为外形可见左耳缺如,右耳发育不全,小眼裂,小下颌,唇腭裂,右侧脐动脉缺失,手畸形,食指重叠于中指上,胎儿孕周宫内生长受限。
Hu等(2020)报道了两个不同的病例,一个例携带母系遗传的18q22.3位点的2.36 Mb微重复,另一个携带父系遗传的18q22.3位点的1.74 Mb微重复。在18q22.3位点有微重复的双亲及其胎儿表型均正常,出生后随访一年,没有发现任何异常。研究者认为18q22.3 (chr18:68,606,012-71,287,101)重复是一种良性变异。
Isidor等(2008)报道一个具有多种先天性畸形的胎儿,表型为不典型的无脑畸形、胼胝体发育不全、小脑发育不全、腭裂、室间隔缺损和主动脉弓发育不全等。胎儿有18q23的450kb重复,遗传自健康的父亲。
18p重复
单纯性18号p部分三体综合征是极其 罕见的,Jacobsen和Mikkelsen于1968年首次报道。至今,文献中报道的单纯性染色体18p部分三体综合征患者有 30余例。
单纯性染色体18p部分三体综合征患者的共有临床表型非常少∶只存在不同程度的智力低下/生长发育迟缓、多变的 面容异常、癫痫等,也有部分携带者表型正常。
邓林贝等(2017)总结了25例文献中报道的单纯性18p部分三体综合征患者的临床表型,如下图。25例已报道的病例中,大部分的共有临床表型包括∶轻到中度智力低下/生长发育迟缓(16/25)、非特征性的面容异常(13/25)、癫痫(4/25)、身材矮小(4/25)和隐睾症(3/25)。
单纯性染色体18p部分三体综合征按照染色体畸变的发生机制可分为4大类∶第一类为常见的非平衡易位型(10/26),即该类患者异常衍生的18号染色体来源于携带18号染色体和非同源染色体平衡易位的父母;第二类为额外的标记染色体型(3/26),即该类患者携带额外的一条标记染色体,该标记染色体来源于18号染色体的部分区域,表现为染色体的数目异常;第三类为复杂的18p缺失合并等臂的18p染色体(del18p+iso18p)型(3/26),该类患者的基因组涉及18号染色体的复杂结构重排,即一条18号染色体短臂缺失合并携带额外的一条等臂18p染色体,该类型一般为新发生的变异,表现为18号染色体的结构合并数目异常;第四类为单纯的18p 重复 型 (dup18p)(10/26),即该类患者的18号染色体结构重排仅发生在其中一条18号染色体短臂上。
wang(2019)等总结了11例单纯性 18p部分三体综合征患者表型(下表),重复区域有18p11.1,18p11.32等, 也可遗传自父母一方,可见该综合征临床表型异质性显著,表型严重程度不一。
18p四体综合征
Tetrasomy 18p syndrome
是一种罕见的染色体疾病。1963年,Froland等首次报道,主要与减数分裂过程中染色体不分离及着丝粒异常分裂导致18 号染色体短臂两个拷贝形成等臂染色体引起,其在新生儿中的发病率约为 1/180 000-1/140 000。 18p 四体的形成通常由于母体第二次减数分裂时染色体的不分离以及着丝粒的异常分离。大多数病例都是生殖细胞形成过程中的随机事件,与母亲的年龄有关,高龄可能影响减数分裂过程所需蛋白等物质的合成,导致同源染色体不分离,或着丝粒分裂异常,从而形成等臂染色体。
18p四体综合征临床表现为 宫内发育迟缓、分娩体重轻,喂养困难,小头畸形,斜视,低耳位,脊柱侧凸,上唇小,手指挛缩偏斜,足为典型的摇摆底,大拇指短,生长发育迟缓,智力障碍,肌张力减退,心脏及肾脏畸形等。
Bawazeer et al (2018)总结18p四体综合征患者表型如下表:
Inan等(2018)报道了一对携带18p四体的同卵双胞胎,但两者在子宫内的表型差异很大,其中1例胎儿系统B超显示存在宫内发育迟缓、脐膨出、脊柱后凸、趾残缺等,而另1例胎儿则在24周前无明显的异常,在24周时超声显示存在生长受限、前鼻骨半透明层增厚、右脚大趾变短、裂开等。
Balasubramanian等(2016)报道了一个家庭中的 父亲(30岁)和 两个女儿(5岁、6岁)均有 18p11.32-p11.31重复(2.6Mb), 他们临床表型为不同程度的智力障碍、语言和精细运动能力发育迟缓、认知/学习能力差、社交和行为困难。
18号染色体臂间倒位
由亲代携带18 号染色体臂间倒位所导致的子代18号染色体非平衡重组病例十分罕见,自1974年Jacobs等首次报道,迄今检索到相关病例报道仅20余例。在以上报道中,先证者大多为出生后确诊,其中既有 表型几近正常者,亦可见出生不久便夭折的婴幼儿。
有研究显示,18号染色体臂间倒位携带者生育非平衡重组子代的概率约为1/3。
对于18号染色体倒位家系的遗传咨询,需告之家系内携带者子代存在发生染色体非平衡重组的可能。对于有生育需求的明确携带者,告之可通过植入前遗传学诊断等方式避免胎儿染色体非平衡重组。对于未行植入前遗传学诊断者,须告之尽管胎儿染色体可通过产前诊断得到确认,然而针对染色体非平衡重组胎儿表型及妊娠结局的预测,目前仍存在较大困难。因此,结合影像检查定期评估胎儿生长发育各项指标,加强随访,充分知情告知,应是针对18号染色体臂间倒位家系提供遗传咨询的基本准则。
参考文献
就不列,很多,如有需要私信作者,如错误请批评指导。返回搜狐,查看更多
责任编辑:
网友评论
最新评论